Shirley ShiDu Yan Lab
 Contact
Contact
Phone: (212) 342-0494
Email: sdy1@cumc.columbia.edu
About
Shirley ShiDu Yan, MD, is a Professor of Surgical Sciences in Surgery. After receiving her medical degree from Fujian Medical College, Dr. Yan completed a postdoctoral fellowship in the Department of Physiology and Cellular Biophysics at Columbia University in New York, USA. Dr. Yan’s research focuses on mitochondrial pathology and molecular and cellular mechanisms of mitochondrial degeneration, cellular stress and survival in neurodegenerative disorders relevant to Alzheimer’s disease, aging, and diabetes. Her research has won multiple awards, including the Dolph C. Simons Award in Biomedical Science, the NIH MERIT Award from the National Institute on Aging, and the Zenith Fellow Award from Alzheimer’s Association. She currently sits on the editorial board for several prominent research publications, including Journal of Alzheimer’s disease. She also severs on research grant review committee for National Institute of health and multiple research foundations. Her research has been supporting by NIH.
Research Focus
Mitochondrial and synaptic degeneration in aging and neurodegenerative diseases, including Alzheimer’s disease; amyloid and tau metabolism, neuroinflammation, aged related metabolites, Alzheimer’s disease mouse model.
Our research focuses on investigating the cellular and molecular mechanism of mitochondrial and neuronal stress and survival in aging and neurodegenerative disorders, including Alzheimer’s disease (AD), Diabetes, and age-related dementia. With the rapid aging of the population, age-related dementia, cognitive decline, and neurodegenerative diseases for which no cures are currently available, have emerged as a tremendous challenge to global public health. Deterioration in mitochondrial structure and function contributes significantly to neurodegeneration in the pathological aging process. Moreover, mitochondrial dysfunction plays an accentuated role in early pathogenesis and therefore constitutes a promising therapeutic target for prevention and early intervention. Thus, understanding the mechanisms and the strategies to rescue mitochondrial and synaptic injury will have a highly significant translational impact for developing effective therapeutics, in particular mitochondrial therapeutics for the treatment and, ultimately, cures of aging-related cognitive decline, neurobehavioral disorder, and dementia.
We have pioneered research to identify the specific cellular and mitochondrial targets (RAGE, receptor for advanced glycation end products, mitochondrial amyloid binding protein, and cyclophilin D) of amyloid-beta peptide (Aβ), and oxidative stress-mediated mitochondrial and neuronal degeneration. We were the first to reveal the progressive accumulation of Aβ in brain mitochondria from human AD patients and transgenic AD mouse models. Early accumulation of Aβ in synaptic mitochondria was associated with mitochondrial dysfunction, which directly linked to synaptic degeneration. My research team was the first to discover the role of cyclophilin D-dependent mitochondrial mPTP in mitochondrial and synaptic dysfunction relevant to the pathogenesis of AD. Our Lab has developed novel transgenic mouse models and ex-vivo AD trans-mitochondrial cytoplasmic hybrid (cybrid) neuronal cells to test the role of these co-factors on neuronal and mitochondrial function. These studies open new avenues for therapeutic intervention for aged- and diabetes-induced dementia, and AD. We are actively investigating mitochondrial alterations linked to synaptic damage and cognitive dysfunction. As shown this diagram, compared to non-AD derived mitochondria (red), AD-derived mitochondria reveal higher levels of ROS, which arrest differentiation and mitochondria movement resulting in less movable mitochondria and more stationary mitochondria (blue) in AD cybrids. Antioxidants suppress ROS accumulation, which in turn rescue mitochondrial defects including axonal mitochondrial transport and neuronal differentiation.
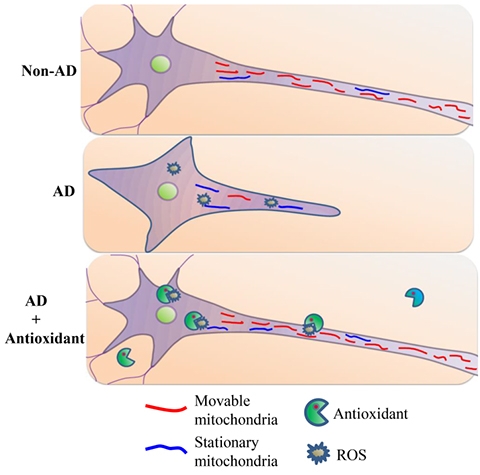
[Image Above] Schematic diagram depicting the role of antioxidant treatment on differentiation ability and axonal mitochondrial transport in AD cybrid cells (Yu, Q et al., JAD 2016)
Mitophagy is a process to remove dysfunctional mitochondria. Thus, impaired mitophagy causes the accumulation of dysfunctional mitochondria and increased reactive oxygen species (ROS). We demonstrate for the first time that decreased PTEN-induced putative kinase 1 (PINK1) expression is associated with AD pathology. Enhancing PINK1-dependent mitophagy signaling striking reduces amyloid pathology, ROS (see images), and mitochondrial and synaptic dysfunction. Conversely, loss of PINK1 activity or blockade of mitophagy signaling fails to reverse amyloid-β-induced detrimental effects. Thus, activation of PINK1 may represent a new therapeutic avenue for combating AD by limiting Aβ pathology and lessening mitochondrial and synaptic injury.
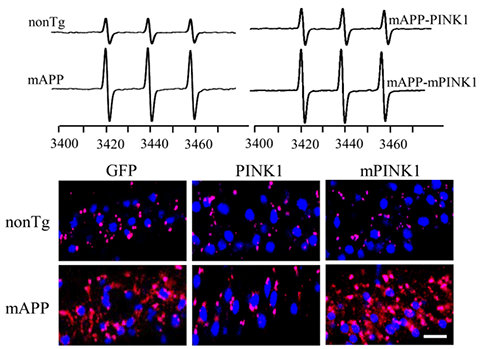
[Image Above] Restoring PINK1 activity suppresses mitochondrial ROS as indicated by MitoSox (red) in brains of AD mice (mAPP). Mutant PINK1 lacking PINK1 kinases activity does not suppress ROS. Blue: nucleus. Top panel, representative EPR spectra in the indicated mice. The peak height in the spectrum indicates levels of ROS. (Fang D et al., Brain, 2017)
Synaptic failure is one of pathological features of AD and nurodegenerative diseases. Synaptic mitochondria are vital for synaptic transmission and plasticity. Our studies elucidate the crosstalk between synaptic mitochondria and synaptic function, such as mitochondrial permeability transition pore (mPTP) and clearance of damaged mitochondrial and toxic metabolites linked to synaptic function. Notably, synaptic stress has reversed detrimental effects on mitochondrial function and production of ROS relevant to AD pathogenesis. We report that elevation of endophilin A1 in Aβ-rich brain leads to change in both long-term potentiation and learning and memory through ROS/p38 mitogen-activated protein (MAP) kinase. We are actively investigating the role of synaptic mitochondria on Aβ and Tau-induced synaptic injury via mitochondrial signaling relevant to mitophagy, mPTP, and mitochondrial degrading enzyme to clear up Aβ.
Aging is a strong risk factor for brain dementia and cognitive decline. Age-related accumulation of toxic metabolites such as advanced glycation end product (AGEs) could serve as danger signals to initiate and accelerate disease process and neurodegenerations. Age-related accumulation of dicarbonyls and AGEs associate with mitochondrial perturbation and oxidative stress. Blocking mitochondrial ROS prevents formation and accumulation of AGEs and AGEs-induced mitochondrial dysfunction. Exogenous AGEs from high AGEs-containing diet, common with high temperature cooking, as in the western world; significantly impair mitochondrial and cognitive function. These studies highlight a novel mechanism by which AGEs-dependent signaling promotes carbonyl stress and sustained mitochondrial dysfunction. Thus, eliminating formation and accumulation of AGEs may represent a new therapeutic avenue for combating cognitive decline and mitochondrial degeneration relevant to aging and neurodegenerative diseases including Alzheimer’s disease. We are actively investigating AGEs-mediated mitochondrial and synaptic perturbation and amyloid and tau pathology.
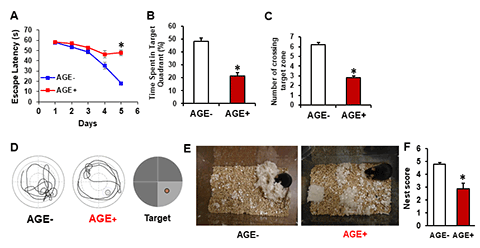
[Image Above] AGE fed mice exhibit impairment in learning and memory and the ability of nest building performance. Mice fed AGE+ diet for 17 months starting at the age of 5 months show longer escape latency during Morris Water Maze (MWM, A) hidden platform task, shorter time spent in the quadrant with the hidden platform (B), and less mean number of crossings of the target during the probe test (C). (D) Representative traces during the probe test showing AGE diet –fed mice impaired finding target. (E) Representative nest photos show that AGE-fed mice are not able to perform the nest building behavior and nesting deficits indicated by incomplete nests and low nest score (F). *P0.01 compared with AGE- fed mouse (Akhter F, et al., JAD, 2020)
Education
- MS: Fujian Medical College (Histopathology and Anatomy), Fujian, China
- MD: Fujian Medical College (Medicine), Fujian, China
Honors
- 2017: Recipient of the Dolph C. Simons, Sr Award in the Biomedical Science in recognition of research achievement in the biomedical sciences to an individual who has had a major and substantial impact and who has been of national and/or international interest.
- 2017: Chair of the section of Molecular & Cell Biology: Metabolic and Other cellular Progresses in neurodegeneration at Alzheimer's Association International Conference 2017 (AAIC), London, the world largest of Alzheimer’s Disease Community.
- 2016-2022: Permanent member of NIH grant review committee for Neural Oxidative Metabolism and Death Study Section, National Institutes of Health
- 2010-2020: NIH MERIT Award from the National Institute on Aging "in recognition of sustained contribution to aging and leadership and commitment to the field." MERIT award is one of the most prestigious awards presented by the NIH and provide long-term support to exceptional outstanding and experienced investigators who contribute significantly to the research field.
- 2018: International Scientific Advisory Board of the 13th International Symposium on the Maillard Reaction (September 10-13, Montreal od Canada)
- 2016: Co-Chair of Cell death and Cell Stress Social, Annual Neuroscience Conference of Society of Neuroscience, San Diego
- 2015: Alzheimer’s research featured at the TechConnect World Conference and Expo in Washington, DC
- 2015: Editor Review Board Member, Current Alzheimer Research
- 2007-2019: Associate Editor, Journal of Alzheimer’s Disease
- 2013-Present: Editorial Advisory Board Member for Current Alzheimer Research
Publications
- Du F, Yu Q, Yan S, Zhang Z, Chen D, Yan SF, and Yan SS. Gain of PITRM1 peptidase in cortical neurons affords protection of mitochondrial and synaptic function in an advanced age mouse model of Alzheimer’s disease. Aging Cell, 2021 May 5;e13368. Doi: 10.111/acel.13368. Online ahead of print
- Du F, Yu Q, and Yan SS. Pink1 activation attenuates impaired neuronal-like differentiation and mitochondrial dysfunction in Alzheimer’s disease trans-mitochondrial cybrid cells. J Alzheimer’s Dis. 2021;81(4):1749-1761. Doi: 10.3233/JAD-210095. Highlighted as a feature on JAD online 8/20/2021
- Akhter F, Chen D, Akhter A, Yan SF, Yan SS. Age-dependent accumulation of dicarbonyls and advanced glycation endproducts (AGEs) associates with mitochondrial stress. Free Radic Biol Med. 2021 Jan 5;164:429-438. doi: 10.1016/j.freeradbiomed.2020.12.021. [Epub ahead of print] PubMed PMID: 33359687. (International Mailard Research Society (IMARS) Highlights of the glycation literature, Editors’ Choice in November 2020-January 2021
- Akhter F, Chen D, Akhter A, Sosunov AA, Chen A, McKhann GM, Yan SF, Yan SS. High Dietary Advanced Glycation End Products impair mitochondrial and cognitive function. J Alzheimer’s Dis. 2020;76(1):165-178. doi: 10.3233/JAD-191236. PubMed PMID: 32444539; PubMed Central PMCID: PMC7581068
- Yu Q, Wang Y, Du F, Yan S, Hu G, Origlia N, Rutigliano G, Sun Q, Yu H, Ainge J, Yan SF, Gunn-Moore F, Yan SS. Overexpression of endophilin A1 exacerbates synaptic alterations in a mouse model of Alzheimer’s disease. Nature Communication, 2018 Jul 30;9(1):2968. doi: 10.1038/s41467-018-04389-0. PMID: 30061577.
- Du F, Yu Q, Chen D, and Yan SS. Astrocyte attenuate mitochondrial dysfunction in human dopaminergic neurons derived from iPSC. Stem Cell Report 2018 Feb 13;10(2):366-374. doi: 10.1016/j.stemcr.2017.12.021. Epub 2018 Jan 27. PMID: 29396183
- Fang F, Yu Q, Arancio O, Chen D, Gore SS, Yan SS, Yan SF. RAGE mediates Aβ accumulation in a mouse model of Alzheimer’s disease via modulation of β- and -secretase activity. Hum Mol Genet. 2018 Jan 10. doi: 10.1093/hmg/ddy017. [Epub ahead of print]
- Du F, Yu Q, Yan S, Hu G, Lue, LF, Douglas W, Tue K, Yan SS. PINK1 signaling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain, 2017 Dec1;140(12):3233-3251. This article has been selected as an Editor's Choice article for the month of December 2017 and was highlighted on our website of the Brain. https://academic.oup.com/brain/article/140/12/3233/4565533?login=true. This article is also highlighted on Nature Review Neurology, Published online 10 Nov 2017: doi:10.1038/nrneurol.2017.160
- Yu Q, Fang D, Swerdlow RH, Yu H, Chen JX, Yan SS. Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J Alzheimers Dis. 2016 Sep6;54(2):679-90. doi: 10.3233/JAD-160532. PMID: 27567872. This paper was selected one of the best publications in JAD in 2016 (Top 20).
- Yan S, Du F, Wu L, Zhang Z, Zhong C, Yu Q, Wang Y, Lue LF, Walker DG, Douglas JT, Yan SS. F1F0 ATP synthase-cyclophilin D interaction contributes to diabetes-induced synaptic dysfunction and cognitive decline. Diabetes. 2016 Nov;65(11):3482-3494, PMID: 2755467.
- Fang D, Qing Y, Yan S, Chen D, Yan SS. Development and Dynamic regulation of mitochondrial network in human midbrain dopaminergic neurons differentiated from iPSCs. Stem Cell Reports. 2016 Oct 11;7(4):678-692. doi: 10.1016/j.stemcr.2016.08.014. PMID: 27666790
- Huang S, Wang Y, Gan X, Fang D, Zhong C, Wu L, Hu G, Sosunov AA, McKhann GM, Yu H, Yan, SS. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes. 2015; 64(5):1728-42. PMID: 25412623, PMCID: PMC4407851
- Du H, Guo L, Yan S, Sosunov AA, Mckhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2010, Oct 26;107(43):18670-5. Epub 2010 Oct Science daily news on Early Role of Mitochondria in Alzheimer's Disease May Help Explain Limitations to Current Beta Amyloid Hypothesis; http://www.sciencedaily.com/releases/2010/10/101013122557.htm
- Du H, Guo L, Fang F, Chen D, Sosunov AA, Mckhann GM, Yan Y, Wang C, Zhang H, Molkentine JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nature Medicine. 2008 Oct;14(10):1097-105. Epub 2008 Sep 21. PMID: 18806802, Faculty 1000 of Biology, (highlighted by Nature Review Drug Discovery 2008, News & Views comments in the same issue of Nature Medicine). This research was ranking top#3 of hot papers in Alzheimer's disease research in year 2008-2010 (432 citations to 1/13/2017)